Naslovnica
Vijesti
Sport
Magazin
Magazin
Shopping
Ljubimci
Food
Mame
Auto
Fit
Chill
Horoskop
Oglasi
Recepti
Ostalo
Zaposli se na Indexu
Lajk.hr
Tečaj
TV program
Kino
Vrijeme
{{userInfo.Initials}}
{{userInfo.FirstName}}
{{userInfo.FirstName}}
Moj profil
Odjavite se
PRIJELOMNA VIJEST
Milanović se obraća.
Vijesti
Hrvatska
Zagreb
Regija
EU
Svijet
Znanost
Crna kronika
Komentari
Novac
Afere
UŽIVO
Milanović: Priprema se državni udar. Ne bojte se, ovo neće proći
PREDSJEDNIK Zoran Milanović obraća se javnosti nakon objave Ustavnog suda kojom mu se brani da bude mandatar, objavivši na Facebooku fotografiju na kojoj predsjednik Ustavnog suda Miroslav Šeparović sjedi s HDZ-ovim ministrima.
Vezane vijesti
VIDEO
Ustavni sud: Milanović ne može biti mandatar ni premijer čak i ako da ostavku
ANKETA
Ustavni sud zabranio Milanoviću da bude mandatar. Slažete li se s odlukom?
Milanović ovom slikom odgovorio Ustavnom sudu
106
Troje ustavnih sudaca: Šeparović prijeti saboru
Izrael napao Iran. U Teheranu prosvjed, ljudi uzvikuju: "Smrt cionistima"
Butković: Zorane, jesi li jučer zvao šefa susjedne države da te spoji s manjincima?
Milanović se obraća. Reakcije na Šeparovića: "Dno dna, nečuveno, zastrašujuće"
Profesor s Prava: Ovo je nečuveno
Tražimo sadržaj koji
bi Vas mogao zanimati
Izdvojeno
Teme i komentari
PARLAMENTARNI IZBORI
Krstulović Opara nakon presice Ustavnog suda: Oporba će opet pozivati na nerede
FOTO
Ovo je dočekalo novinare na Pantovčaku
HDZ-ovac koji je dobio manje glasova od Ave: Ljevica je poražena
RIJEKE PRAVDE NA IZBORIMA
Matija Posavec: Očito je da Ustavni sud ima dvostruke kriterije
Siniša Hajdaš Dončić kaže da je nezadovoljan rezultatom SDP-a
Ivan Penava: Zasad nemam komentara o Ustavnom sudu
HDZ NA IZBORIMA
Marijana Puljak: Velika je panika u HDZ-u, koriste Ustavni sud da im spasi kožu
Sandra Benčić za Index: Izgleda kao da Ustavni sud igra za HDZ
SDP-ovci: Neki od oporbenjaka se pretvaraju u HDZ-ove žetončiće
Najnovije
Najčitanije
1
min
UŽIVO
Milanović: Priprema se državni udar. Ne bojte se, ovo neće proći
2
min
Izrael napao Iran. U Teheranu prosvjed, ljudi uzvikuju: "Smrt cionistima"
4
min
Milanović se obraća. Reakcije na Šeparovića: "Dno dna, nečuveno, zastrašujuće"
10
min
Muškarci podmetali požare u Crikvenici, nastala šteta od 250.000 eura
10
min
Srpski medij: Brat osumnjičenog za ubojstvo Danke je umro nasilnom smrću
Prikaži još vijesti
1
Milanović se obraća. Reakcije na Šeparovića: "Dno dna, nečuveno, zastrašujuće"
2
Izrael napao Iran. U Teheranu prosvjed, ljudi uzvikuju: "Smrt cionistima"
3
VIDEO
Ustavni sud: Milanović ne može biti mandatar ni premijer čak i ako da ostavku
4
Milanović ovom slikom odgovorio Ustavnom sudu
5
Grmoja: Imamo dovoljno ruku. Most i DP bez Možemo i SDSS-a
Prikaži još vijesti
Sport
Nogomet
Transferi
Košarka
Tenis
Regija
Komentari
Borilački sportovi
Ostali sportovi
Bočkaj se vraća u Dinamo?
ISTIČE mu posudba.
Vezane vijesti
Pogledajte golčinu Petra Bočkaja iz slobodnog udarca
Bočkaj se oprostio od Dinama: Dva naslova prvaka ću uvijek pamtiti
Bočkaj ima novi klub
7
Ovo je najluđa liga. Četiri kluba u igri za naslov, a igrat će se još šest derbija
Redknapp ispričao anegdotu o Modriću: Svi su me upozoravali da će biti "ubijen"
Hrvatska nakon velikog preokreta dobila novog predstavnika na Olimpijskim igrama
Pred ispadanjem su u treću ligu, a sljedeće sezone bi mogli igrati Europa ligu
Guardiola o najboljem strijelcu PL-a: Što smo mogli napraviti?! Sam je htio otići
Tražimo sadržaj koji
bi Vas mogao zanimati
Izdvojeno
Teme i komentari
HNL
Trener Gorice: Ako pobijedimo Rijeku, vodim ekipu na benzinsku ili na janje
Što će biti s Jakirovićevim otkrićem? Proigrao je, a Bayern bi spustio cijenu
Bio je prvak s Hajdukom, a sad će voditi momčad protiv njega. Ovako je najavio susret
LIGA PRVAKA
Govorili su im da više nisu liga Petice. Sada su na korak do 6 klubova u Ligi prvaka
Modrić plakao nakon drame sa Cityjem? Španjolci otkrili što je rekao članovima Reala
Ancelottijev sin je ključan u Realovoj svlačionici. Španjolci otkrili njegove zadaće
INDEX SPORT INTERVJU
Junak s Poljuda za Index: San mi je igrati za hrvatsku reprezentaciju
"Takav se rodi jednom u ne znam koliko godina. Zato je najbolji sportaš Hrvatske"
U Hrvatskoj mi sude zbog imena i prezimena, a ona je moja koliko i njihova
Najnovije
Najčitanije
19
min
Prodaje se klub koji vodi Ivan Leko
46
min
Hrvatska nakon velikog preokreta dobila novog predstavnika na Olimpijskim igrama
48
min
Ovako je Mourinho prije pet godina pričao o Xabiju Alonsu
49
min
Guardiola o najboljem strijelcu PL-a: Što smo mogli napraviti?! Sam je htio otići
51
min
Bočkaj se vraća u Dinamo?
Prikaži još vijesti
1
Livaković je već pao kod izvođenja penala. Onda je izveo čudo
2
Casemira pitali tko je najbolji: Modrić, Kroos, on ili Barcine legende? Ovo je rekao
3
Dvojica izlaze pred Livakovića. Pogledajte što radi hrvatski junak turskog velikana
4
Govorili su im da više nisu liga Petice. Sada su na korak do 6 klubova u Ligi prvaka
5
Modrić plakao nakon drame sa Cityjem? Španjolci otkrili što je rekao članovima Reala
Prikaži još vijesti
Magazin
TV & Film
Glazba
Tech & Gadget
Lifestyle
Showbiz
Zanimljivosti
UŽIVO
Pogledajte kakvo je iznenađenje dočekalo Zagrepčane koji su ušli u tramvaj 12
DUBIOZA Kolektiv iznenadila je Zagrepčane.
Vezane vijesti
Dubioza Kolektiv i Dino Šaran u novoj pjesmi: "Ne palite TV jer sve može otić U3PM"
Dva albuma Dubioze Kolektiva konačno su na vinilu
1
10 najfatalnijih uloga Hollywooda. Prvo mjesto je nedodirljivo
OGLAS
Rock & Roll Z brega na dol napokon u Zagrebu
FOTO
Ovako su danas izgledale Bačvice
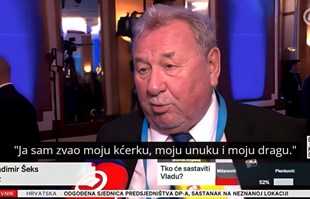
Novinarka pitala Šeksa tko će sastaviti vladu, njegov odgovor postao viralan
Hrvati se sprdaju s novom političkom dramom u državi, pogledajte što pišu
Tražimo sadržaj koji
bi Vas mogao zanimati
Izdvojeno
Teme i komentari
KNJIGE
Pronašli smo pet domaćih romana koje vrijedi pročitati
Približava se Noć knjige, knjigu fizičara koji je rasprodao Lisinski morate nabaviti
Travanjski hitovi u domaćim knjižarama: Bestseler koji se čita na 28 jezika
TEME HRVOJA MARJANOVIĆA
10 činjenica o kultnim glazbenim hitovima koje sigurno niste znali
Pogledali smo Fallout. Oduševit će čak i one koji nisu gejmeri
Najbizarniji predizborni plakati u Hrvatskoj od početka 90-ih nadalje
TAYLOR SWIFT
Osveta nakon 8 godina: Taylor Swift u naslovu pjesme sakrila ime Kim Kardashian?
Zbog albuma koji je izašao prije nekoliko sati nastao je kaos na društvenim mrežama
Sutra izlazi album godine. Milijuni još od veljače odbrojavaju dane do njega
Najnovije
Najčitanije
7
min
UŽIVO
Pogledajte kakvo je iznenađenje dočekalo Zagrepčane koji su ušli u tramvaj 12
12
min
FOTO
Ovako su danas izgledale Bačvice
32
min
Edda ima dvije godine i život ju nije mazio. Možete li ju udomiti?
37
min
10 najfatalnijih uloga Hollywooda. Prvo mjesto je nedodirljivo
41
min
Otkriveno tko će zamijeniti Melkiora i Damira u žiriju MasterChefa
Prikaži još vijesti
1
Ekonomistice, studentice... Tko su cure koje se natječu za Miss Universe Hrvatske?
2
Salma Hayek i njena pokćerka privukle poglede u Italiji, nosile haljine sa šljokicama
3
Za tetovažom Plenkovića nastala pomama. Vlasnik salona: Moram pripremiti više verzija
4
Zbog albuma koji je izašao prije nekoliko sati nastao je kaos na društvenim mrežama
5
Melkior i Damir odlaze iz žirija MasterChefa, otkrili su razloge
Prikaži još vijesti
1h15m
Veganski mramorni kuglof na šalice
10m
....i malo Tonke
Šlag torta od 4 sastojka
1h40m
Mate Janković
Rolade od svinjskih leđa s dinstanim aromatiziranim kiselim kupusom
1h
Krem juha od krumpira i boba
1h50m
Birilica
Marzipan stollen
Automobili
36.390
Nekretnine
157.319
Prodaje se golf 6 gti
13.000 €
Vespa LXV
800 €
Mercedes Viano
13.000 €
Žena iz BiH komentira muža kako je zapeo u rijeci dok peca, video je svjetski hit
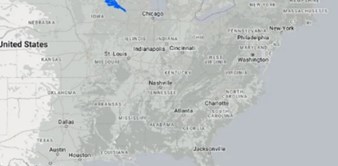
Netko je napravio mapu koja pokazuje kako bi izgledalo kad bi se Hrvatska preselila u SAD
Svi imaju samo jedno pitanje kad vide ovu fotku iz Bosne: Zašto?
15 zbunjujućih fotki zbog kojih bi vam se mogli posvađati razum i oči
Shopping
Vidi sve
Hailey Bieber popularizira nošenje marame u bakinom stilu
G-Shock predstavlja novi GravityMaster sat. Dolazi u tri boje
Cowgirl stil sada je prisutan i u uređenju domova
Vizažistica Blake Lively otkrila da ova hidratantna krema čini kožu iznimno glatkom
Food
Vidi sve
Kava bez kofeina potencijalno bi mogla biti nesigurna za konzumaciju
Melkior i Damir odlaze iz žirija MasterChefa, otkrili su razloge
U Poreču održani 15. Svijet malvazija i 30. Ocjenjivanje vina, ovo su pobjednici
Istarska maslinova ulja osvojila tri zlata na prestižnom ocjenjivanju u New Yorku
Mame
Vidi sve
Mama pokazala što sve zna njezin 10-mjesečni sin, ljudi su zapanjeni
U omiljenom dječjem crtiću prvi put spomenut LGBTQ+ par. Mnogi roditelji oduševljeni
Tata pokazao kako izgledaju igraonice u javnom prijevozu u Finskoj
Roditelji probušili uši tromjesečnoj bebi pa razbjesnili javnost: “Ali zašto?”
Ljubimci
Vidi sve
Edda ima dvije godine i život ju nije mazio. Možete li ju udomiti?
U potrazi ste za ljubimcem? Ovi slatkiši traže zauvijek dom
Prvi put nabavljate kućnog ljubimca? Evo šest ključnih savjeta
Vlasnica upoznala pse s novim ljubimcima, pogledajte preslatki susret
Fit
Vidi sve
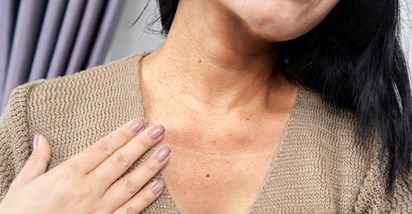
Suptilna promjena na koži mogla bi biti rani pokazatelj tihog ubojice
Ove namirnice pomažu mozgu koji stari, a nisu dio mediteranske prehrane
Tri razloga zašto je vježbanje s utezima ključno za mršavljenje
Evo koje zanimanje najviše doprinosi odličnom pamćenju u starosti
Auto
Vidi sve
Mercedes odustao od ideje malenog dvocilindraša u S klasi
Prodaja električnih automobila u Europi u padu
VIDEO
Potpuno nova Mazda CX-80 apsolutno pripada premium klasi
Ove kineske automobile dizajnirao je ex-dizajner Alfa Romea i Lamborghinija
Chill
Vidi sve
Ljudi se slažu da je ovaj klasik najpotresniji film koji su ikad gledali
Pronašli smo pet domaćih romana koje vrijedi pročitati
Psihološki triler s Bruceom Willisom šesti je najgledaniji film na Netflixu
Evo zašto je Jack Nicholson uhićen na premijeri filma 1968.
Horoskop
Vidi sve
Nadolazeći dani bit će posebno teški za dva horoskopska znaka
Vodenjake danas čekaju uzbudljivi razgovori, Bikovi bi mogli dobiti bonus
U ovim znakovima rađaju se žene koje muškarce osvajaju bez ikakvog truda
Horoskopski znakovi koji su uvjereni da se cijeli svijet vrti oko njih
Prijavite se
Brza prijava
(traje par sekundi)
Facebook
Google
Prijava emailom
Ako već niste registirani, poslat ćemo vam email s linkom za nastavak registracije.
Prijavite se
Kreiranjem i prijavom na račun slažete se s našim
Uvjetima korištenja
i
Pravilima o privatnosti



































































Uvjetima korištenja i Pravilima o privatnosti